Publication Summaries
15. Qibin Geng, Yushun Wan, Fu-Chun Hsueh, Jian Shang, Gang Ye, Fan Bu, Morgan Herbst, Rowan Wilkens, Bin Liu, Fang Li (2023) Lys417 acts as a molecular switch that regulates the conformation of SARS-CoV-2 spike protein eLife https://doi.org/10.7554/eLife.74060
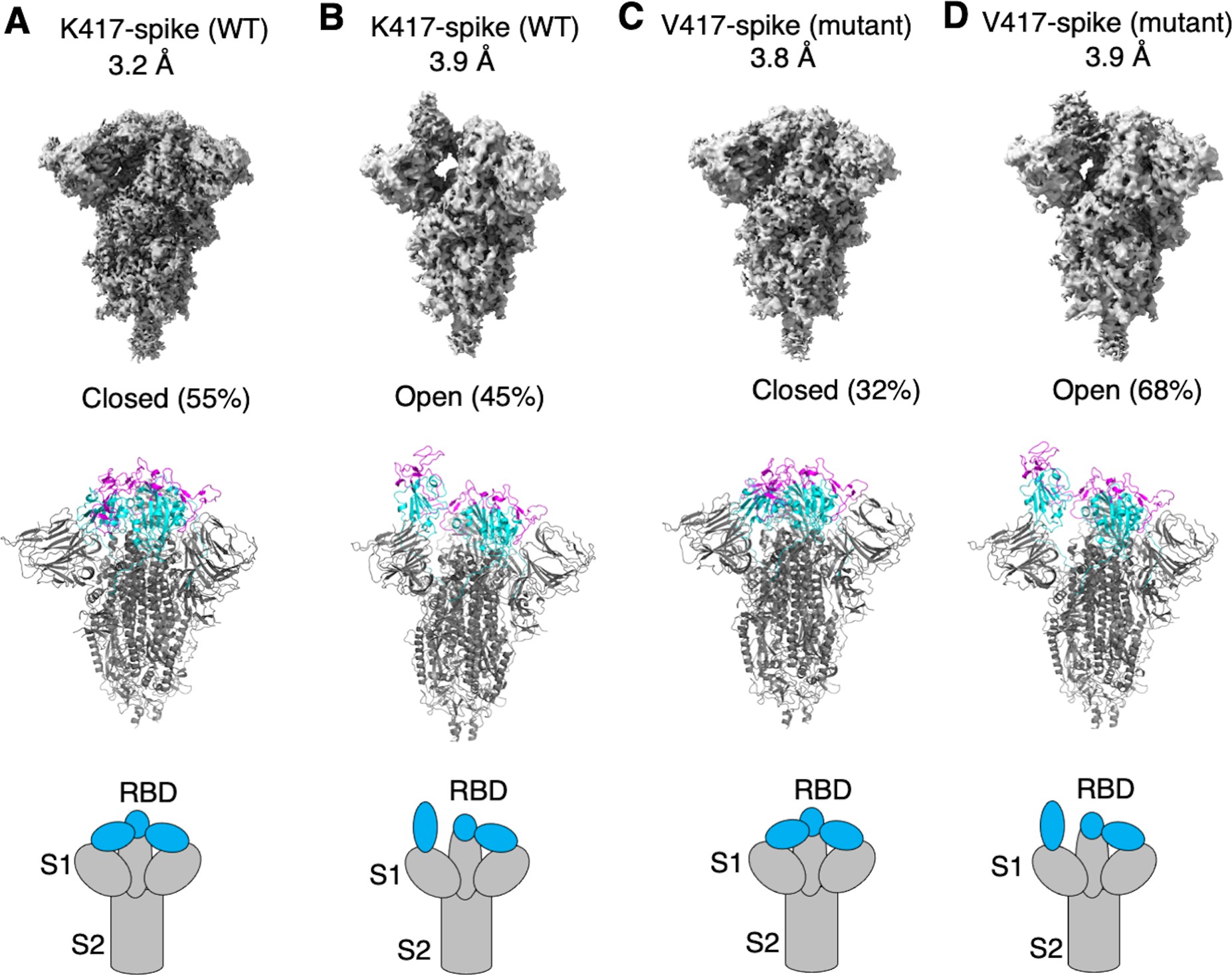
SARS-CoV-2 spike protein plays a key role in mediating viral entry and inducing host immune responses. It can adopt either an open or closed conformation based on the position of its receptor-binding domain (RBD). It is yet unclear what causes these conformational changes or how they influence the spike’s functions. Here, we show that Lys417 in the RBD plays dual roles in the spike’s structure: it stabilizes the closed conformation of the trimeric spike by mediating inter-spike–subunit interactions; it also directly interacts with ACE2 receptor. Hence, a K417V mutation has opposing effects on the spike’s function: it opens up the spike for better ACE2 binding while weakening the RBD’s direct binding to ACE2. The net outcomes of this mutation are to allow the spike to bind ACE2 with higher probability and mediate viral entry more efficiently, but become more exposed to neutralizing antibodies. Given that residue 417 has been a viral mutational hotspot, SARS-CoV-2 may have been evolving to strike a balance between infection potency and immune evasion, contributing to its pandemic spread.
14. (2023) The ever-expanding diversity and complexity of the Arenaviridae family, Virulence, 14:1, DOI: 10.1080/21505594.2023.2279353
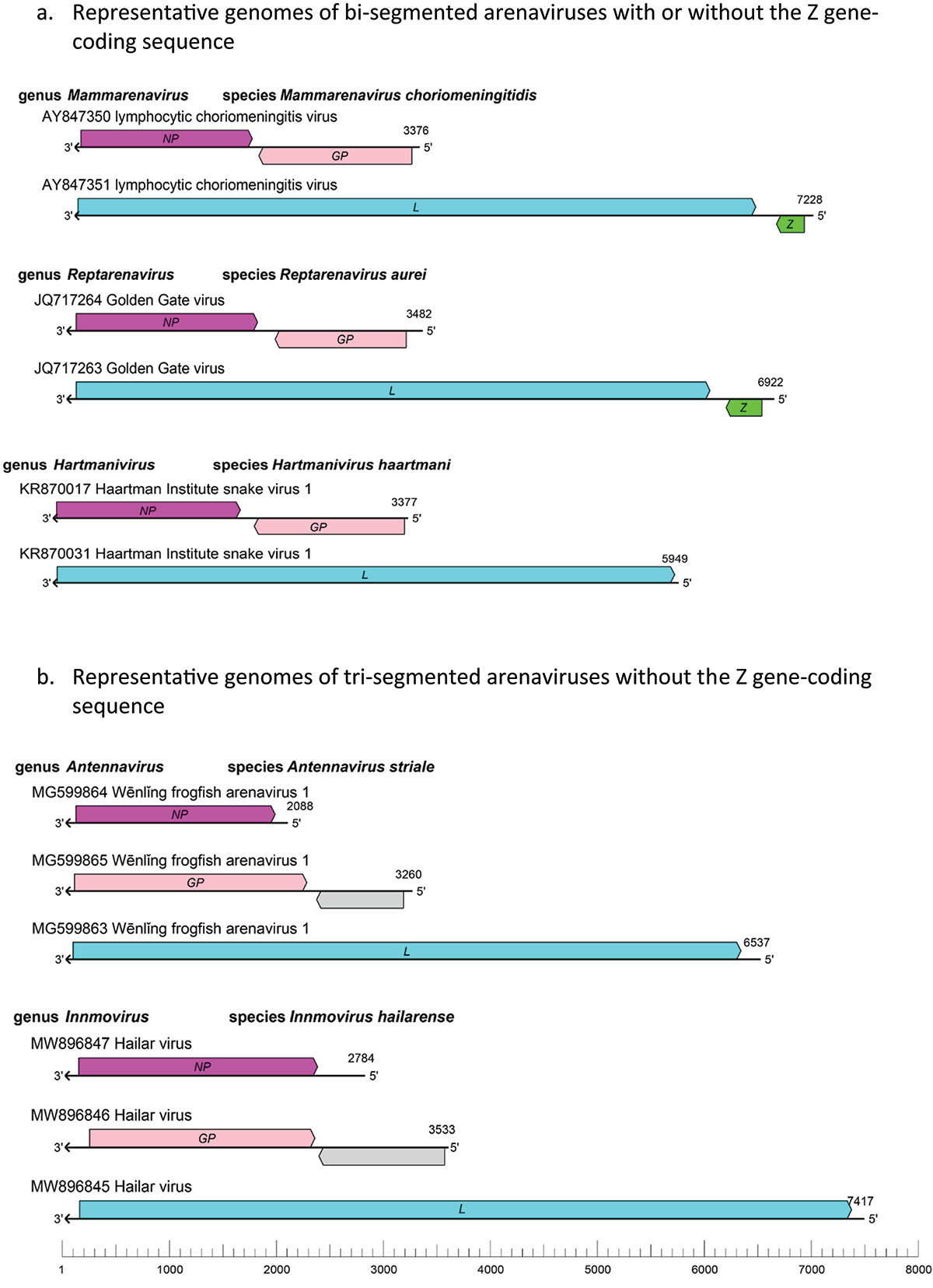
Viruses are generally classified as DNA or RNA viruses based on the type of nucleic acids that make up their genomes. DNA viruses, in general, were phylogenetically more similar in genomic sequences and host specific than RNA viruses [Citation1]. An example of RNA viruses with increasingly diverse genomic architectures and host species is the Arenaviridae family. This editorial article aims to highlight similarities and differences of known and newly discovered arenaviruses, their expanding host ranges, and the diseases that they can cause. As some of these viruses can cause severe and sometimes fatal diseases in humans, it is important to keep track of them to prevent potential future outbreaks.
13. Ye G, Pan R, Bu F, Zheng J, Mendoza A, Wen W, Du L, Spiller B, Wadzinski BE, Liu B, Perlman S, Li F. Discovery of Nanosota-2, -3, and -4 as super potent and broad-spectrum therapeutic nanobody candidates against COVID-19. J Virol. 2023 Oct 19:e0144823. doi: 10.1128/jvi.01448-23. PMID: 37855638. https://journals.asm.org/doi/10.1128/jvi.01448-23
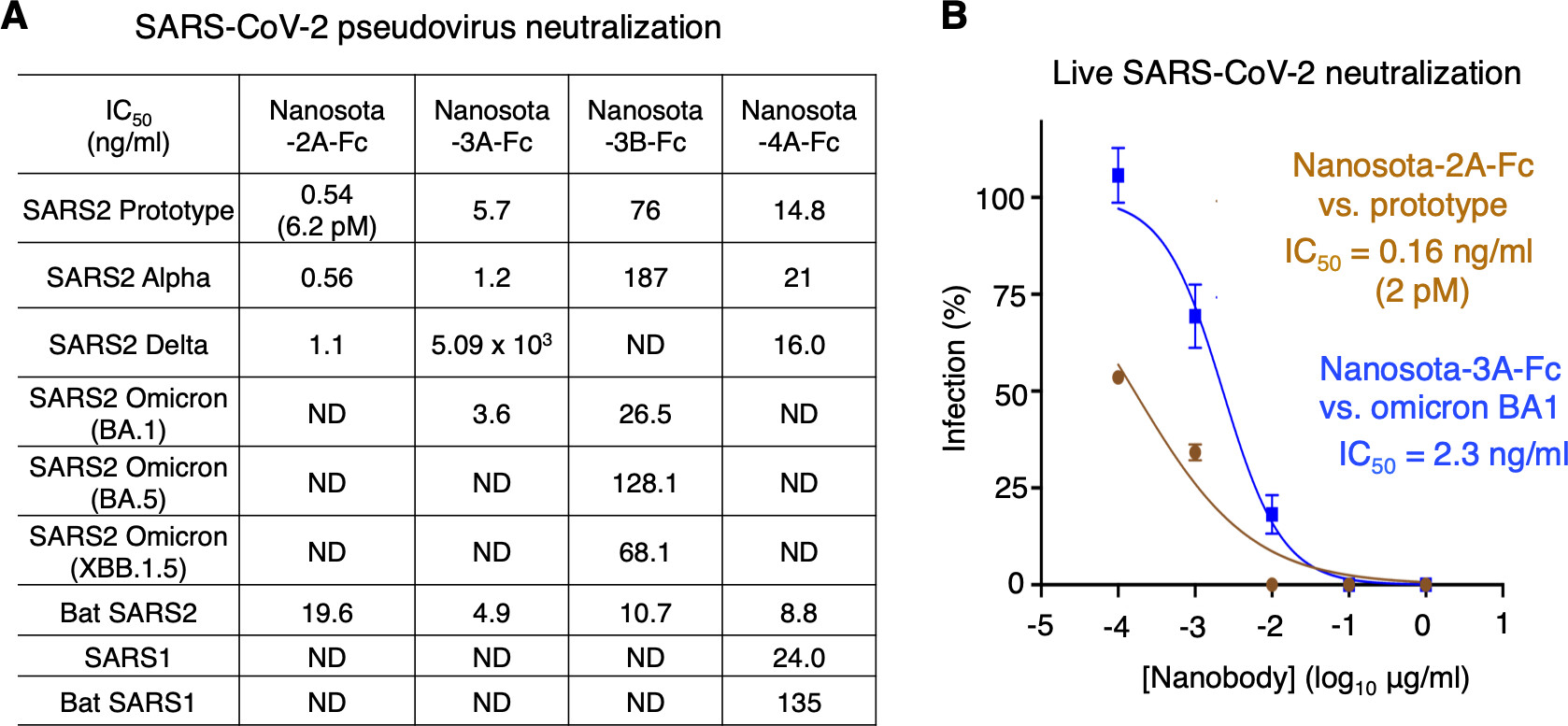
Nanobodies are single-domain antibodies derived from camelid animals. Here, we discovered three anti-SARS-CoV-2 nanobodies, namely, Nanosota-2, -3, and -4, from an alpaca immunized with SARS-CoV spike protein. We further characterized the antiviral activities of these Fc-tag-fused nanobodies. Notably, Nanosota-2 inhibits the prototypic SARS-CoV-2 strain in vitro (with an IC50 of 2 pM) and in mice (at a dosage of 4 mg/kg or administered 18 hours post-challenge). These potency metrics are the best among known SARS-CoV-2 entry inhibitors.
Moreover, Nanosota-3 effectively inhibits the omicron variant, both in vitro and in mice, regardless of the administration route (intraperitoneal or intranasal). Furthermore, Nanosota-3 has been biochemically engineered to inhibit both early and currently circulating subvariants of omicron. Additionally, Nanosota-4 uniquely inhibits both SARS-CoV-1 and SARS-CoV-2. Cryo-EM data revealed that the three nanobodies bind to functionally critical and non-overlapping regions in the spike protein. Given their cost-effectiveness, ease of adaptation to new viral strains, and potential use as inhalers, the Nanosota series are powerful therapeutic tools against coronavirus pandemics.
12. Chongsaritsinsuk, J., Steigmeyer, A.D., Mahoney, K.E. et al. Glycoproteomic landscape and structural dynamics of TIM family immune checkpoints enabled by mucinase SmE. Nat Commun 14, 6169 (2023). https://doi.org/10.1038/s41467-023-41756-y
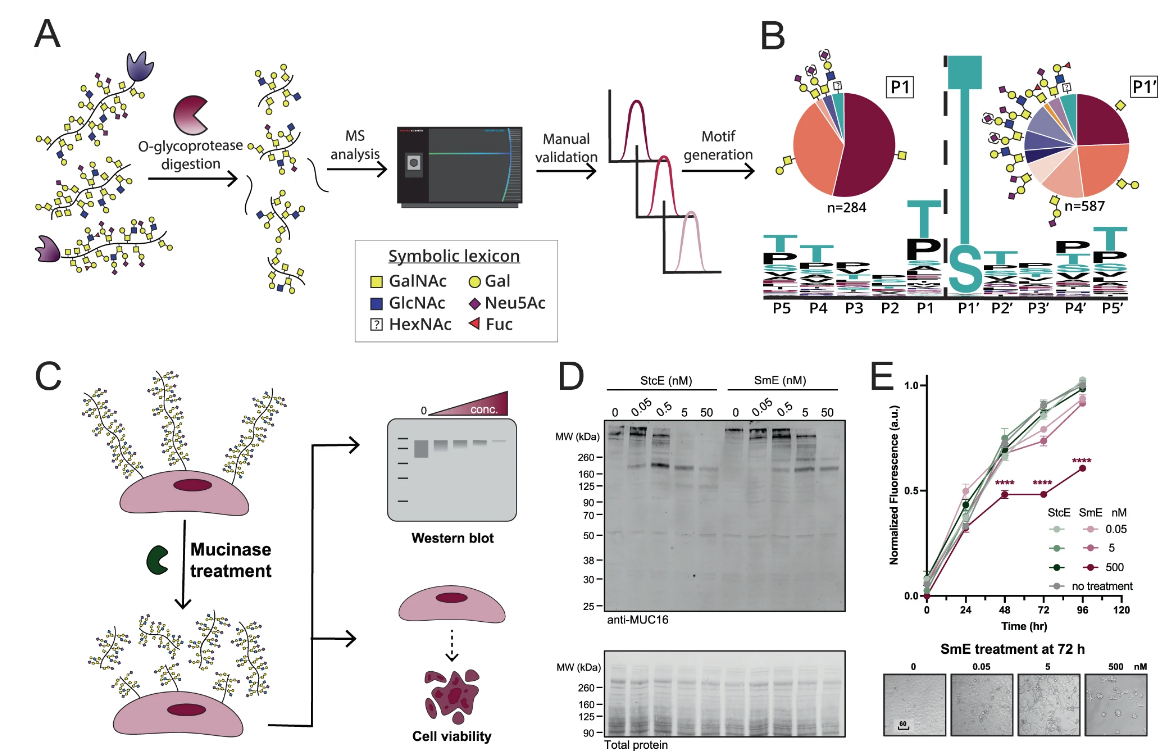
Mucin-domain glycoproteins are densely O-glycosylated and play critical roles in a host of biological functions. In particular, the T cell immunoglobulin and mucin-domain containing family of proteins (TIM-1, -3, -4) decorate immune cells and act as key regulators in cellular immunity. However, their dense O-glycosylation remains enigmatic, primarily due to the challenges associated with studying mucin domains. Here, we demonstrate that the mucinase SmE has a unique ability to cleave at residues bearing very complex glycans. SmE enables improved mass spectrometric analysis of several mucins, including the entire TIM family. With this information in-hand, we perform molecular dynamics (MD) simulations of TIM-3 and -4 to understand how glycosylation affects structural features of these proteins. Finally, we use these models to investigate the functional relevance of glycosylation for TIM-3 function and ligand binding. Overall, we present a powerful workflow to better understand the detailed molecular structures and functions of the mucinome.
11. Jimmidi, R., Chamakuri, S., Lu, S. et al. DNA-encoded chemical libraries yield non-covalent and non-peptidic SARS-CoV-2 main protease inhibitors. Communications Chemistry 6, 164 (2023). https://doi.org/10.1038/s42004-023-00961-y
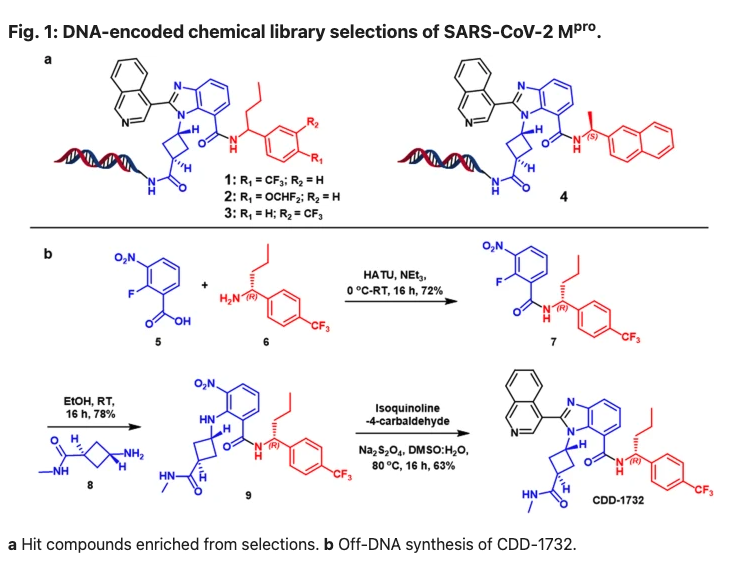
Conventional structure-based design of Mpro inhibitors of SARS-CoV-2 often starts from the structural information of Mpro and their binders; however, the continual rise of resistant strains requires innovative routes to discover new inhibitors. Here, the authors develop a DNA-encoded chemical library screening to produce non-covalent, non-peptidic small molecule inhibitors for SARS-CoV-2 Mpro independently of preliminary knowledge regarding suitable starting points.
Read the full paper here: https://www.nature.com/articles/s42004-023-00961-y
10. Kim, S. H., Kearns, F. L., Rosenfeld, M. A., Votapka, L., Casalino, L., Papanikolas, M., Amaro, R. E., & Freeman, R. (2023). SARS-CoV-2 evolved variants optimize binding to cellular glycocalyx. Cell reports. Physical science, 4(4), 101346. https://doi.org/10.1016/j.xcrp.2023.101346
Viral variants of concern continue to arise for SARS-CoV-2, stymieing detection efforts like testing and impacting the understanding of a variant’s mechanisms of action. This paper investigates the effect of an evolving spike protein in SARS-CoV-2 variants and subsequent interactions. The positively charged Omicron variant evolved enhanced binding rates to the negatively charged glycocalyx. Moreover, the authors discovered that while the Omicron spike-ACE2 affinity is comparable to that of the Delta variant, the Omicron spike interactions create a significantly enhanced bound to ACE2.
The findings suggest that SARS-CoV-2 variants evolve to be more dependent on viral attachment and infection. This discovery enables researchers to engineer a second-generation test strip to reliably detect all concern variants, including Omicron.
9. Moghadasi, S. A., Heilmann, E., Khalil, A., Nnabuife, C., Kearns, F., Ye, C., Moraes, S. N., Costacurta, F., Esler, M., Aihara, H., von Laer, D., Martinez-Sobrido, L., Palzkill, T., Amaro, R. E., & Harris, R. (2022). Transmissible SARS-CoV-2 variants with resistance to clinical protease inhibitors. Science Advances, 2023-03-31. https://doi.org/10.1101/2022.08.07.503099
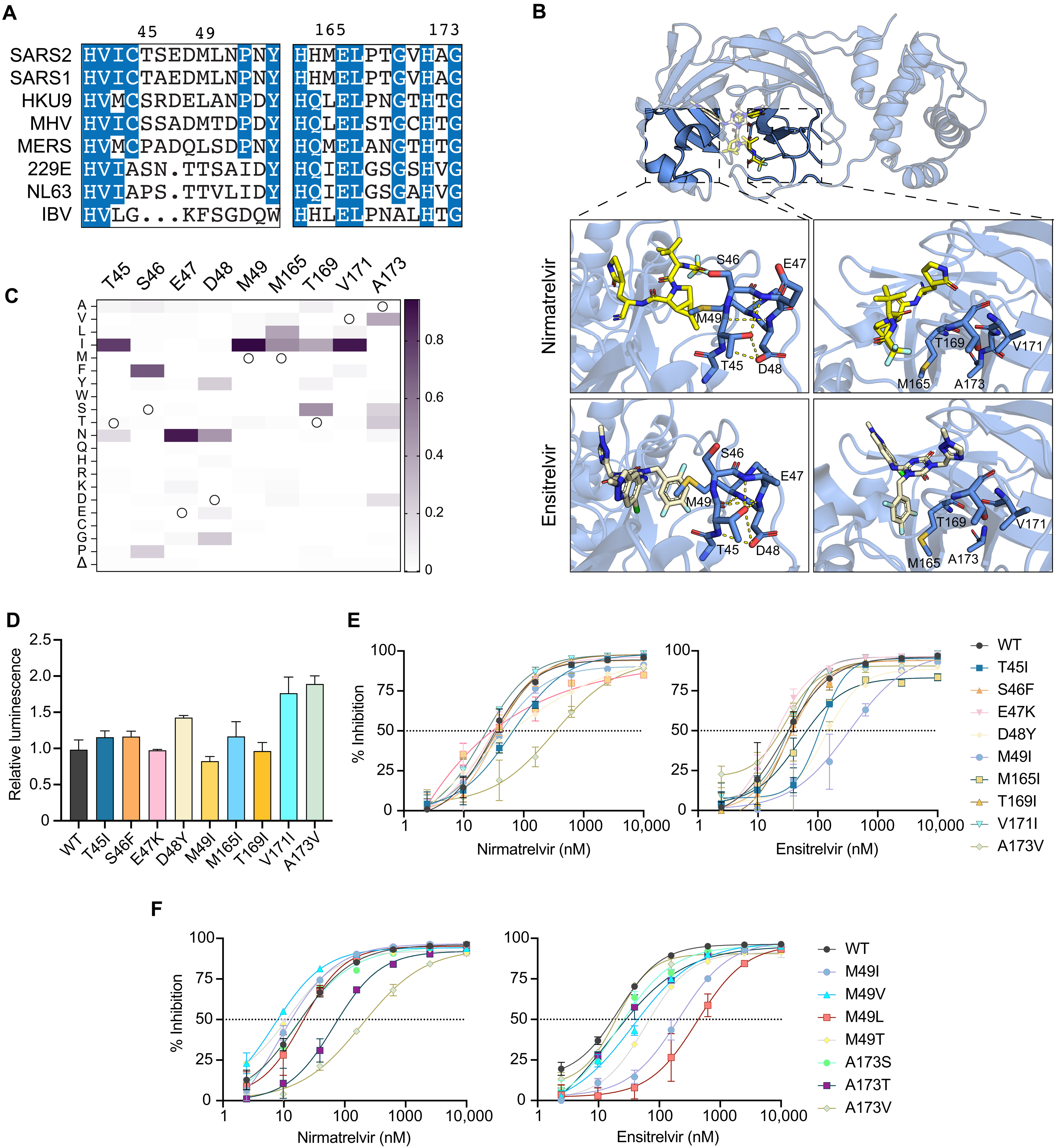
Variable active site residues elicit differential resistance to nirmatrelvir and ensitrelvir.
Vaccines and drugs have helped reduce disease severity of SARS-CoV-2. However, ongoing virus transmission, continuous evolution, and increasing selective pressures have the potential to yield viral variants capable of resisting current vaccines and drugs. This paper investigates the susceptibility of natural variants of the main protease (Mpro/3CLpro) of SARS-CoV-2 to protease inhibitors. Multiple single amino acid changes in Mpro confer resistance to nirmatrelvir (the active component of Paxlovid). An additional clinical-stage inhibitor, ensitrelvir (Xocova), shows a different resistance mutation profile.
These results encourage monitoring resistance variants and the development of additional protease inhibitors and other antiviral drugs with different mechanisms of action and resistance profiles for therapy.
8. Seyed Arad Moghadasi, Rayhan G. Biswas, Daniel A. Harki, Reuben S. Harris, Rapid resistance profiling of SARS-CoV-2 protease inhibitors, bioRxiv: the preprint server for biology doi: https://doi.org/10.1101/2023.02.25.530000
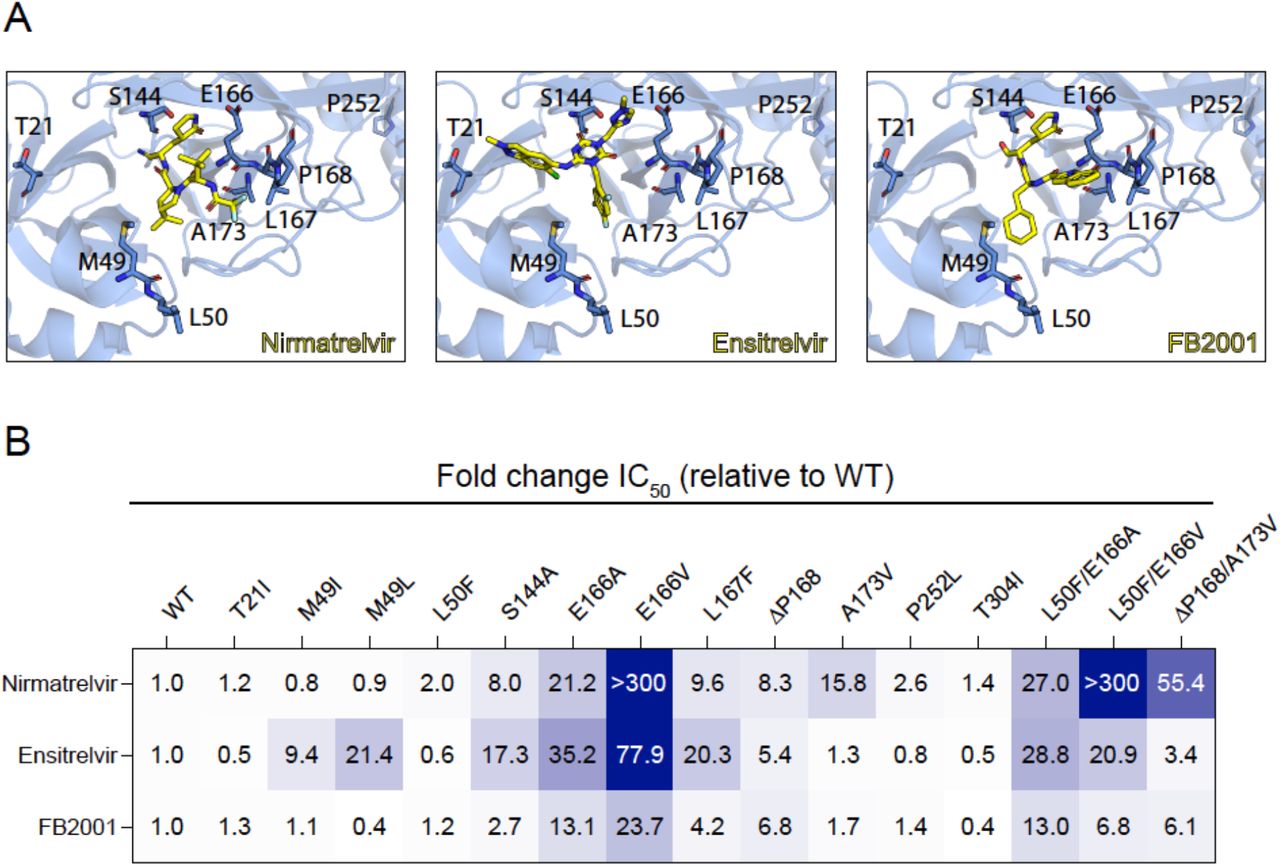
Resistance profiles of nirmatrelvir, ensitrelvir, and FB2001.
Resistance to nirmatrelvir (Paxlovid) has been shown by multiple groups and may already exist in clinical SARS-CoV-2 isolates. Here a panel of SARS-CoV-2 main protease (Mpro) variants and a robust cell-based assay are used to compare the resistance profiles of nirmatrelvir, ensitrelvir, and FB2001. The results reveal distinct resistance mechanisms (“fingerprints”) and indicate that these next-generation drugs have the potential to be effective against nirmatrelvir-resistant variants and vice versa.
7. Smith E, Davis-Gardner ME, Garcia-Ordonez RD, Nguyen TT, Hull M, Chen E, Yu X, Bannister TD, Baillargeon P, Scampavia L, Griffin P, Farzan M, Spicer TP. High throughput screening for drugs that inhibit 3C-like protease in SARS-CoV-2. SLAS Discov. 2023 Apr;28(3):95-101. doi: 10.1016/j.slasd.2023.01.001. Epub 2023 Jan 14. PMID: 36646172; PMCID: PMC9839384. https://pubmed.ncbi.nlm.nih.gov/36646172/
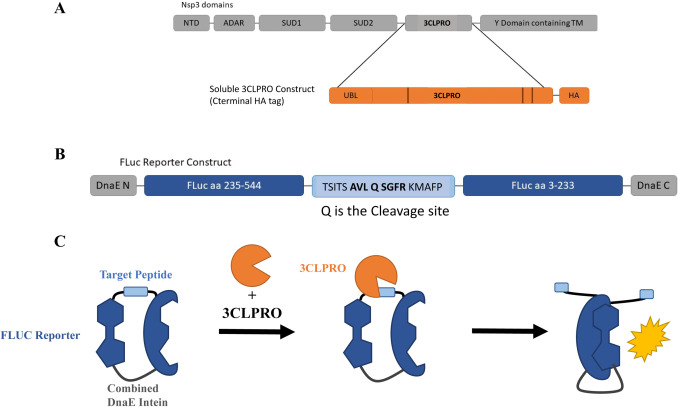
The high rate of variant evolution makes clear the need for new therapeutics that can be clinically applied to minimize or eliminate the effects of COVID-19. With a hurdle of 10 years, on average, for first-in-class small molecule therapeutics to achieve FDA approval, the fastest way to identify therapeutics is by drug repurposing.
To this end, we developed a high throughput cell-based screen that incorporates the essential viral 3C-like protease and its peptide cleavage site into a luciferase complementation assay to evaluate the efficacy of known drugs encompassing approximately 15,000 clinical-stage or FDA-approved small molecules. Confirmed inhibitors were also tested to determine their cytotoxic properties. Medicinal chemistry efforts to optimize the hits identified Tranilast as a potential lead. Here, we report the rapid screening and identification of potentially relevant drugs that exhibit selective inhibition of the SARS-CoV-2 viral 3C-like protease.
6. Kaïn van den Elsen, Bing Liang Alvin Chew, Jun Sheng Ho, Dahai Luo, Flavivirus nonstructural proteins and replication complexes as antiviral drug targets, Current Opinion in Virology, Volume 59, 2023, 101305, https://doi.org/10.1016/j.coviro.2023.101305.

Many flaviviruses are well-known pathogens, such as dengue, Zika, Japanese encephalitis, and yellow fever viruses. Among them, dengue viruses cause global epidemics and threaten billions of people. Effective vaccines and antivirals are in desperate need. In this review, we focus on the recent advances in understanding viral nonstructural (NS) proteins as antiviral drug targets. We highlight a few well-characterized inhibitors targeting these NS proteins and provide an update about the latest development.
Studies aiming to elucidate the architecture and molecular basis of viral replication will offer new opportunities for novel antiviral discovery. Direct-acting agents against dengue and other pathogenic flaviviruses may be available very soon.
5. Heilmann, E., Costacurta, F., Moghadasi, S. A., Ye, C., Pavan, M., Bassani, D., Volland, A., Ascher, C., Weiss, A. K. H., Bante, D., Harris, R. S., Moro, S., Rupp, B., Martinez-Sobrido, L., & von Laer, D. (2023). SARS-CoV-2 3CLpro mutations selected in a VSV-based system confer resistance to nirmatrelvir, ensitrelvir, and GC376. Science translational medicine, 15(678), eabq7360. https://doi.org/10.1126/scitranslmed.abq7360
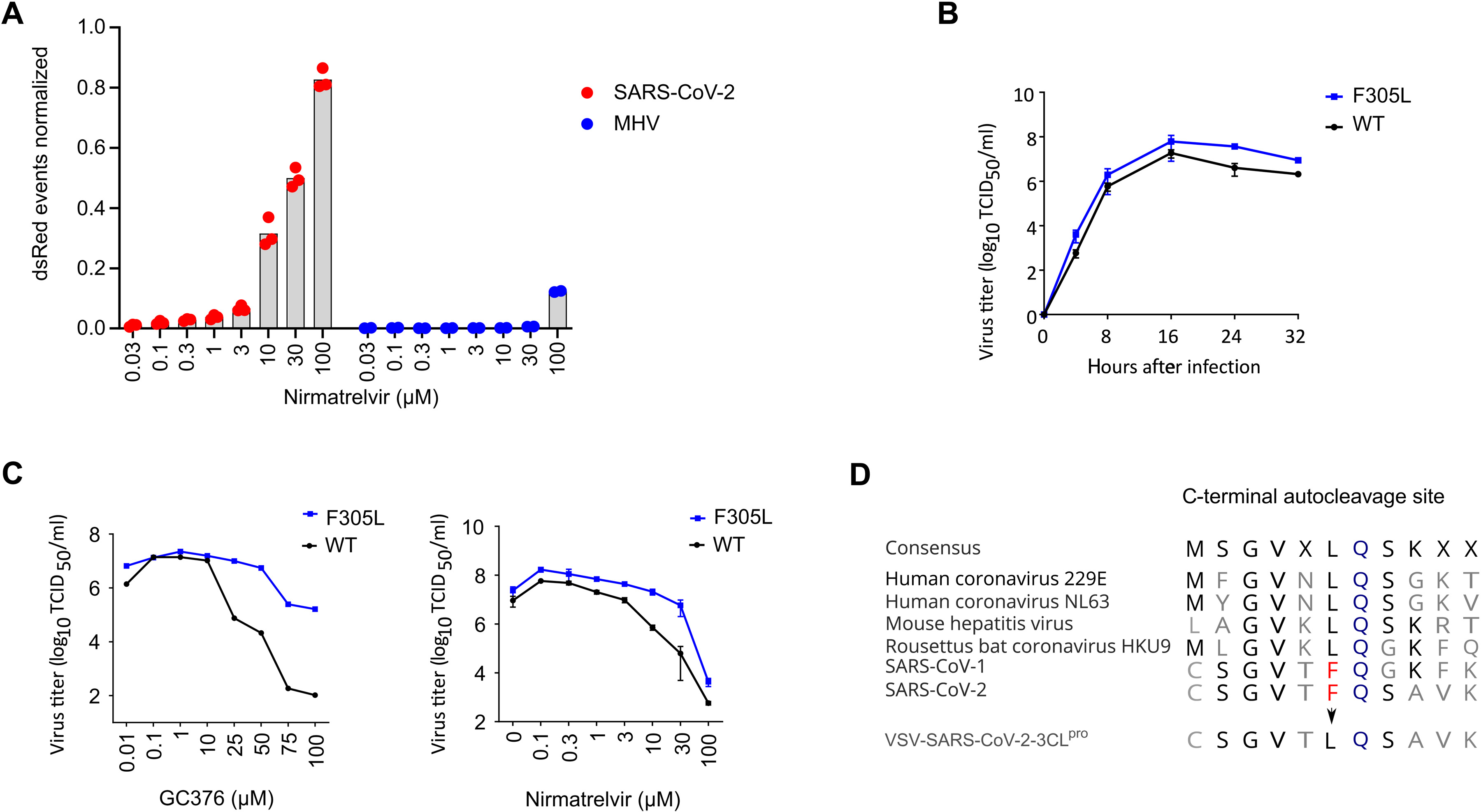
A VSV-based non–gain-of-function system was developed to predict SARS-CoV-2 3CLpro mutations.
Nirmatrelvir, ensitrelvir, and GC376 are SARS-CoV-2 3CLpro inhibitors that provide a promising strategy for treating individuals with COVID-19. However, as antivirals become more widely used, SARS-CoV-2 may develop mutations that create resistance against these antivirals.
The authors used a system to select for nirmaltrelvir resistance mutations, identifying several. Some of the mutations were specific to nirmaltrelvir, whereas others conferred resistance to additional 3CLpro inhibitors.
4. Murphy, H., & Ly, H. (2022). Understanding Immune Responses to Lassa Virus Infection and to Its Candidate Vaccines. Vaccines, 10(10), 1668. https://doi.org/10.3390/vaccines10101668
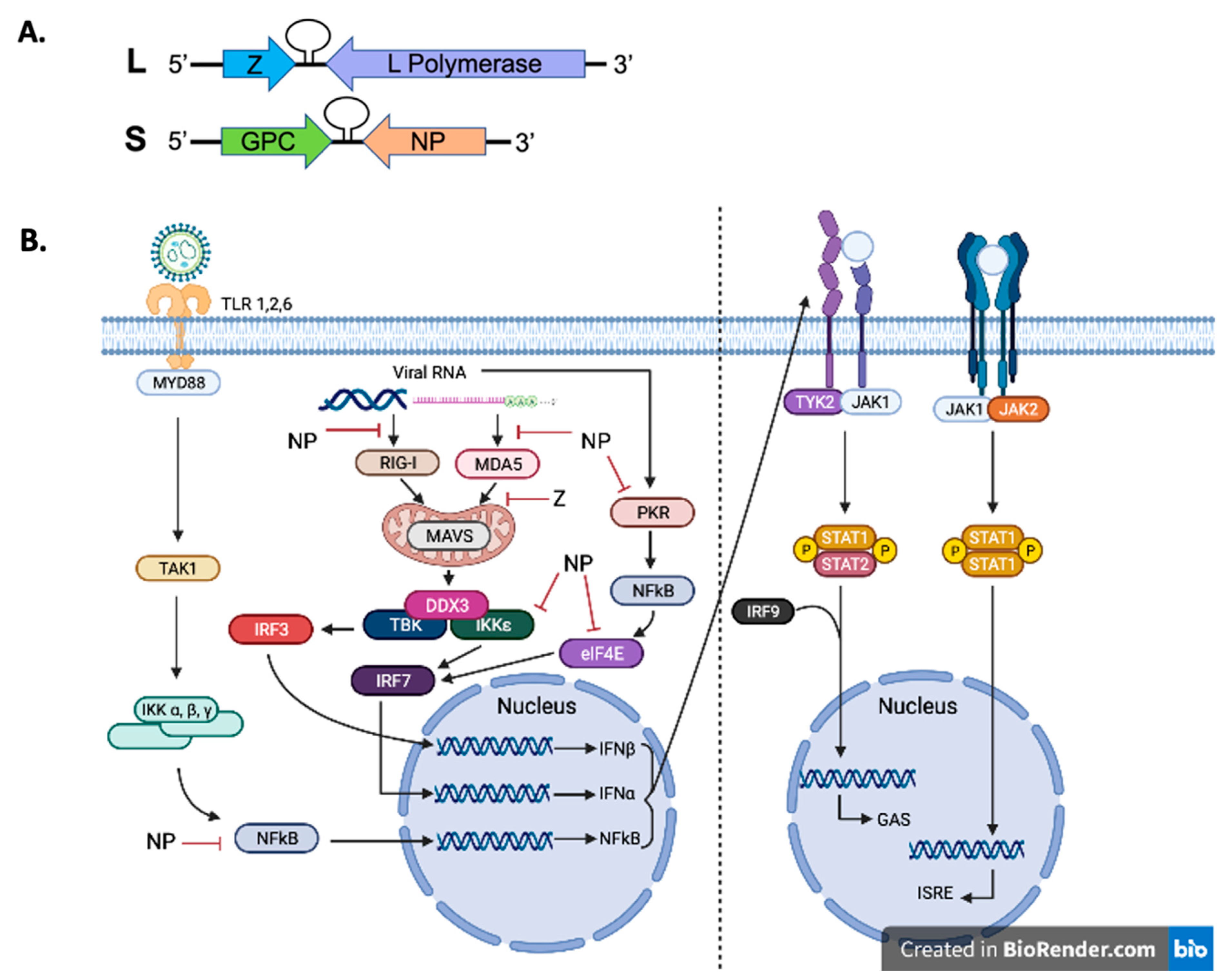
Arenaviral RNA genome structure and inhibition of innate immunity.
Lassa fever (LF) is a deadly viral hemorrhagic fever disease that is endemic in several countries in West Africa. It is caused by Lassa virus (LASV), which has been estimated to be responsible for approximately 300,000 infections and 5,000 deaths annually. LASV is a highly pathogenic human pathogen without effective therapeutics or FDA-approved vaccines. Here, we aim to provide a literature review of the current understanding of the basic mechanism of immune responses to LASV infection in animal models and patients, as well as to several of its candidate vaccines.
3. Moeller, N. H., Passow, K. T., Harki, D. A., & Aihara, H. (2022). SARS-CoV-2 nsp14 Exoribonuclease Removes the Natural Antiviral 3'-Deoxy-3', 4'-dihydro-cytidine Nucleotide from RNA. Viruses, 14(8), 1790. https://doi.org/10.3390/v14081790
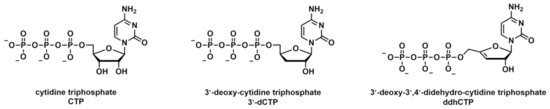
Chemical structures of CTP, and its chain-terminating analogues, 3′-dCTP and ddhCTP.
SARS-CoV-2 features a proofreading mechanism to facilitate the replication of its large RNA genome.
Here we show biochemically that SARS-CoV-2 nsp14 can excise the natural antiviral chain-terminating nucleotide. These results suggest that nsp14 ExoN could play a role in protecting SARS-CoV-2 from natural antiviral activity, which is produced as part of the innate immune response against viral infections, and that the SARS-CoV-2 enzymes may have adapted to minimize the antiviral effects.
2. Kearns, F. L., Sandoval, D. R., Casalino, L., Clausen, T. M., Rosenfeld, M. A., Spliid, C. B., Amaro, R. E., & Esko, J. D. (2022). Spike-heparan sulfate interactions in SARS-CoV-2 infection. Current opinion in structural biology, 76, 102439. https://doi.org/10.1016/j.sbi.2022.102439
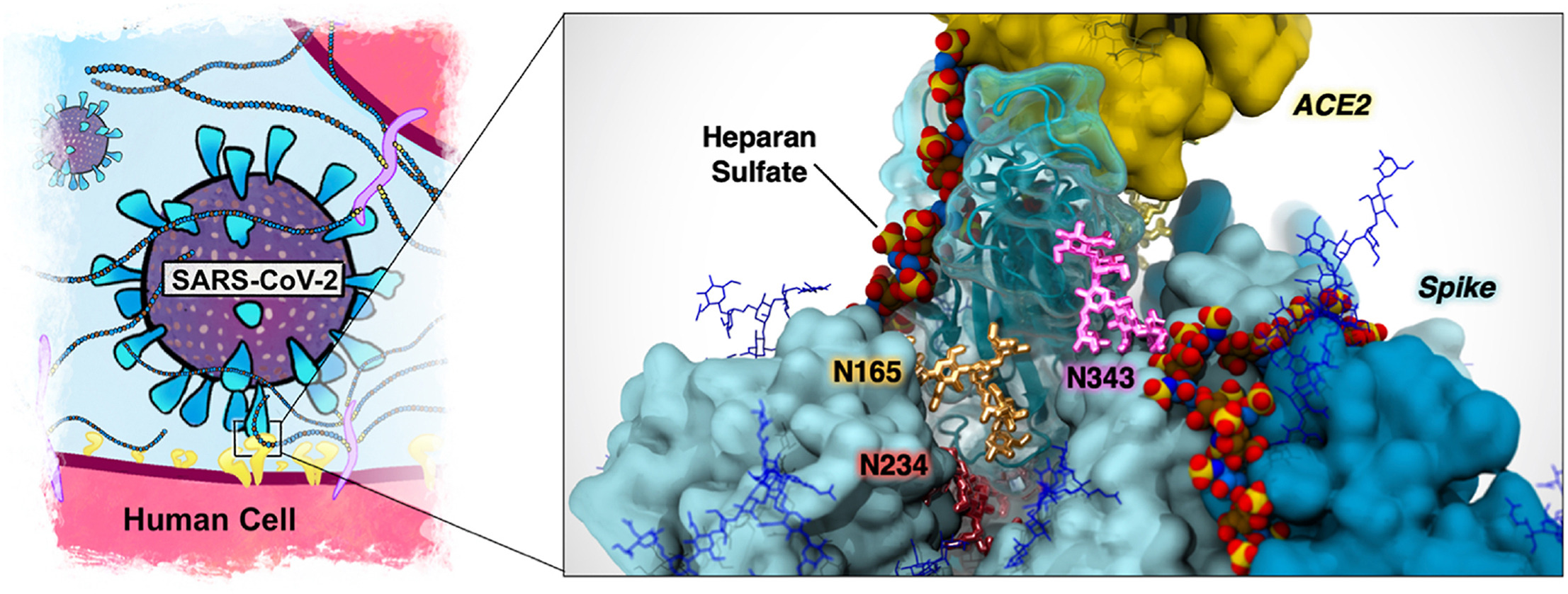
SARS-CoV-2 is a single-stranded RNA, positive-sense, enveloped virus that primarily targets the respiratory epithelial cells lining the upper and lower airways with evidence of spread to other organs. The spike (S) protein studs the outer membrane of the virus and mediates the initial steps of viral infection and spread. It acts by engaging cellular host receptors and it undergoes processing via the transmembrane protease serine 2 or other proteases.
Additionally, infection is facilitated by heparan sulfate (HS), a long negatively charged polysaccharide expressed by all animal cells. Here we review current literature demonstrating the involvement of HS in SARS-CoV-2 infection. We discuss the mechanism of action from a structural perspective and explore the coordination between HS binding and asparagine-linked glycans.
1. Yang, Y., & Du, L. (2022). Neutralizing antibodies and their cocktails against SARS-CoV-2 Omicron and other circulating variants. Cellular & molecular immunology, 19(8), 962–964. https://doi.org/10.1038/s41423-022-00890-1
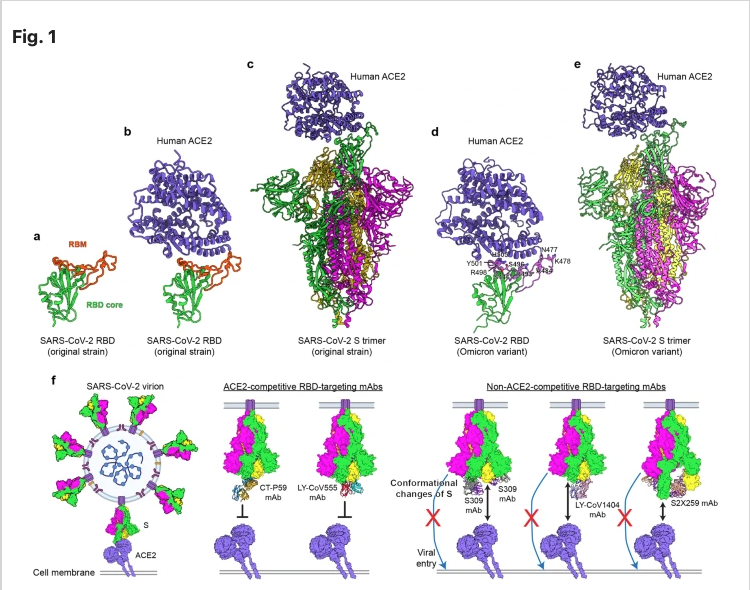
Receptor recognition and cell entry mediated by the SARS-CoV-2 spike (S) protein and its inhibition by neutralizing antibodies.
Compared with the original SARS-CoV-2, the Omicron variant carries more mutations than any other variant identified thus far, among which approximately 39 and 15 substitutions are within the S protein and Receptor Binding Domain (RBD). Crystal and cryo-electron microscopy (cryo-EM) structures of Omicron S/RBD-ACE2 complexes demonstrate that the Omicron S trimer harbors substitutions at a number of RBD residues on the outer surface, with upright RBD(s) being responsible for receptor binding.
However, these mutations in the RBD do not significantly reduce the binding affinity of the RBD for the ACE2 receptor. Many neutralizing monoclonal antibodies (mAbs) were developed based on the original SARS-CoV-2 strain with the aim of preventing and treating SARS-CoV-2 infection. Therefore, it is important to understand whether these mAbs neutralize SARS-CoV-2 variants and whether antibody cocktail treatments retain neutralizing activity.